

Before we go to study the mechanism of action of drugs, it
is important that we understand the concept of pharmacokinetics and pharmacodynamics.
What is pharmacokinetic of a drug?
The easiest way to remember what pharmacokinetics refers to
is to think of it in terms of what the
body does to a drug. So let's think about it, we either swallow a tablet
or apply a cream on our skin. The first thing that takes place is absorption so
the drug has to absorb. Once it gets
absorbed either through the skin or through the stomach it gets into your
bloodstream and then from there it gets distributed into the fluids outside and
inside the cells. Once the drug gets distributed all over the body the body
starts metabolizing, it basically modifies the drug so that it is easy to
excrete. This is done primarily by a liver but, it can also be done by other
tissues so for simplicity drug passes through liver gets bio transformed and
finally it gets eliminated. So elimination is the last step in which drug and
its metabolites get excreted primarily in bile urine and feces.
Now let's
quickly recap what we learned about pharmacokinetics
Pharmacokinetics involves four basic steps:
- First drug has to get absorbed. (Absorption)
- Secondly, once it reaches the systemic circulation, it gets distributed outside and inside the cells. (Distribution)
- Then it starts to get metabolized. (Metabolism)
- Liver plays an important role in that finally drug gets eliminated. (Elimination)
Now let's talk about them in a little bit more detail
There are many routes by which we can administer a drug such as parenteral topical nasal rectal, etc., but unless the drug is given Intravenous it must cross some membrane before it gets into systemic circulation. The absorption of drugs can happen in four different ways
- First through passive diffusion.
- Secondly, through facilitated diffusion.
- Thirdly, through active transport.
- Finally, through endocytosis
What is Passive drug diffusion?
The most of drugs are absorbed by passive diffusion, in
passive diffusion drugs simply move from areas of high concentration to an area
of lower concentration. If it is a water-soluble molecule, it will easily move
through a channel or a pore that's in the membrane now on the other hand, if it
is lipid soluble it will just easily pass through a membrane without any help.
What is Facilitated drug diffusion?
Some other drugs, especially larger molecules will pass
with the help of carrier proteins. Just like in passive diffusion they also move from areas of high concentration to an area
of low concentration and the only difference is that
they actually need a little bit of help from the carrier proteins that are in
the membrane.
What is Active drug transport?
Some drugs transported across the membrane via active
energy dependent transport, unlike passive and facilitated diffusion energy for
this process is derived from ATP. When ATP undergoes hydrolysis to ADP, there
is a high energy that comes from breaking of phosphate bond.
What is Drug Endocytosis?
In endocytosis, drugs of very large size get transported
via engulfment by cell membrane because of their large size they wouldn't fit
in a channel or a pocket of a carrier protein.
NOTE: We also need to remember that absorption is
not exactly that straight forward, it is a variable process depending on pH
surface area and blood flow and this also leads us to a concept of
bioavailability.
 |
| Absorption of drugs |
Let me ask you a question?
If you take a 100 milligram oral
tablet how much of it actually gets absorbed in unchanged form?
The answer is it's not a 100 percent, this is because
unlike drug given intravenously oral medication gets metabolized in gut and in
the liver and good portion of it gets cleared out before it reaches systemic circulation.
Once we administer the drug either orally or intravenously we can measure the
plasma drug concentration over time.
What does AUC mean?
A drug given IV
would start at a concentration of 100 percent because it bypasses the whole
absorption process. However a drug given orally would have to get absorbed
first and then some of it would get eliminated before it even reaches systemic
circulation. Therefore, its curve would look a little different. Once we can
graph this phenomenon we can then find areas
under these curves also known as the AUC.

AUC is really helpful in making comparisons between formulations
and routes of administration, finally knowing all that bioavailability is
simply AUC for the oral drug over AUC for the IV drug times 100. Once the drug
gets absorbed it then gets distributed from circulation to the tissues
What factors affect distribution of a drug?
The distribution process is dependent on a few different factors such as
Lipophilicity: Highly lipophilic drug will dissolve through
some membrane much easier than the hydrophilic drug.
Blood
flow: Some organs such as brain
receive more blood flow than other organs like for example skin. If a drug can
pass through the blood-brain barrier, it will accumulate much faster in the
brain as opposed to in the skin.
Capillary
permeability: For instance
capillaries in the liver have lots of slit junctions through which large
proteins can pass, on the other hand, in the brain, there are no slit junctions
at all so it is more difficult for a drug to pass through.
Binding to
plasma proteins and tissues: Due
to their chemical properties some drugs will accumulate in some tissues more
than the others, also many drugs will bind to albumin, which is a major drug
binding protein that will significantly slow the distribution process
Finally we need to factor
in the volume of distribution which is the theoretical volume that the drug
would have to occupy in order to produce the concentration that's present in
blood plasma.
What is the significance of Volume of distribution?
Volume of distribution can be calculated by taking the amount of drug in the body and dividing it by concentration of the drug in blood plasma

For example, high molecular weight drugs tend to be
extensively protein bound and don't pass through the capillaries as easily as
smaller molecules. Thus, they have higher concentration in blood plasma and
lower volume of distribution typically opposite is true for lower molecular
weight drugs, especially the lipophilic ones which will distribute extensively
into tissues and will result in a larger volume of distribution.
The volume of distribution helps predict whether the drug
will concentrate largely in the blood or in the tissue, this is really helpful
in estimating drug dosing. For example, if drug has a large volume of
distribution, we would need to administer a larger dose to achieve the desired
concentration.
What is Elimination in
Pharmacokinetics?
The last step in the pharmacokinetics process is elimination which refers to clearing of a drug from the body mainly through hepatic renal and biliary route. The total body clearance is simply the sum of individual clearance processes.
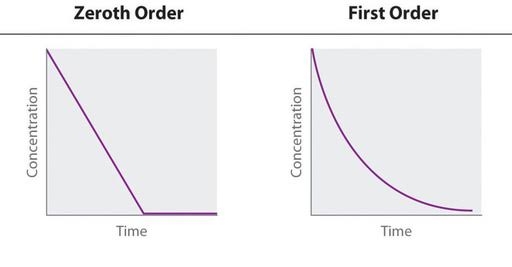
FIRST ORDER KINETICS
Most of drugs are
eliminated by first order kinetics which means that the amount of the drug
eliminated over time is directly proportional to the concentration of
drug in the body.
For example,
starting with 1000 milligrams of a drug the amount eliminated per each time
period will be different but the fraction
will be constant. In this example
per each time period constant of 16 percent of a drug gets eliminated, however
the milligram amount changes and if we were to collect these samples and plot
them the graph would produce a curve that looks something like this
1000 mg → 840 mg → 706 mg → 593 mg (-16 % of drug eliminated)
ZERO
ORDER KINETICS:
There are few drugs such as Aspirin that are eliminated by
zero order kinetics which means that the amount of drug eliminated is independent of drug concentration in the body. So
the rate of elimination is constant.
If we were to take 1000 milligrams again as an example this
time amount of drug eliminated is the same per each time period, which is 200
milligrams but the fraction the
percentage is different and if we were to graph it the zero order
elimination would produce a straight line also the cool thing about these
graphs is that if we can plot them it's easy to determine half-life of a drug
from them.
1000 mg (-20%) →
800 mg (-25%) → 600 mg (-33%) → 400 mg
HALF-LIFE:
Half-life is simply
the time that is required to the reduce
drug concentration in plasma by a half. It can tell us a lot about duration of
action of a drug. Half-life also helps us predict steady state concentrations.
When doses of a drug are repeatedly administered a drug
will accumulate in the body until the rate of administration equals the rate of
elimination this is called steady state.
If we were to graph it when after each additional dose the
peak and trough concentrations stay the same, reached steady state. This is typically
attained in about 4 to 5 half-lives.
Why we are interested in steady
state?
The reason why we are interested in steady state is because
we want concentration of a drug high enough to be effective but not too high to
be toxic. The goal is to maintain steady state concentration within therapeutic
range. Now there are situations such as life-threatening infections during
which we can't waste time getting to steady-state. To compensate for the accumulation time large loading dose can be
administered on treatment initiation to reach desired concentration more rapidly.
NOTE: The most important route of elimination is
through kidney which excrete drugs into the urine. However a kidney can't
efficiently get rid of lipid soluble drugs as there a passively reabsorbed and
that is where the liver comes to the rescue by transforming lipophilic drugs
into water soluble substances that are then easily removed by the kidneys.
Liver accomplishes that mainly through two metabolic
reactions called Phase 1 & Phase 2
Phase 1 reactions
are all about making a drug more hydrophilic these reactions involve introduction
or unmasking of a polar functional group. In Phase 1 we are going to see oxidation
hydrolysis and reduction. It’s also important to remember that most of
these reactions are catalyzed by cytochrome
p450 enzymes.
If metabolites from phase 1 are still too lipophilic, they
can undergo conjugation reaction which involves the addition of a polar group
and this is what happens in phase 2.
In phase 2 we are going to see glutathione conjugation acetylation sulfation and glucuronidation. These reactions produce polar conjugates which cannot diffuse across membranes, therefore they are easily eliminated from the body.
Cytochrome p450 this
large family of enzymes is essential for the metabolism of drugs and there are
few that are worth remembering because they catalyze vast majority of phase 1 reactions
and these are CYP 3A4/5, CYP 2D6, CYP 2C8/9 and CYP 1A2.
Many drug interactions arise from the drug's ability to induce or inhibit these
enzymes.
Some of the
important inducers include Phenytoin, Carbamazepine,
Rifampin, Alcohol (with chronic use), Barbiturates and St. John's Wort , we
can use to remember as "PCRABS".
Some of the important inhibitors are Grapefruit, Protease inhibitors, Azole antifungals, Cimetidine Macrolides
(with exception of Azithromycin), Amiodarone and Non -dihydropyridine calcium channel
blockers (Diltiazem and Verapamil) and again you can use to remember as
"GPACMAN"




){kind=link}
0 Comments
Please do not enter any spam link in the comment box.